Aaron Newman, associate professor of biomedical data science, has been awarded a Trailblazer Cancer Research Grant by the American Association for Cancer Research. The grant supports early-stage and mid-career investigators with $1 million over three years to support innovative, paradigm-shifting cancer research projects. A mid-career investigator awardee, Newman will focus on real-time profiling of tumor microenvironment dynamics to decode immunotherapy response in melanoma.
Next Frontier in Medicine: AI That Learns, Adapts, and Helps Develop Medical Cures
Computational scientist and keynote speaker John Marioni believes artificial intelligence (AI) has become so powerful it is now able to make real-world discoveries using publicly available medical and biological data—in turn helping develop new and better drugs faster than ever before.
Addressing the first annual Computational Biology and AI Research Symposium held by the Stanford Medicine Department of Biomedical Data Science, Marioni noted that targeting a specific molecule in a disease and developing a drug typically takes over eight years, with only a 1% success rate. However, the integration of AI presents exciting prospects for generative AI to accelerate this lengthy process and potentially double the success rate of bringing life-saving drugs to market.
“It’s the next technology that’s going to be transformative,” Marioni, senior vice president and global head of computational sciences at Genentech, said of generative AI. “So, it’s great that Stanford has this program here and we have the ability to talk about it today.”
Marioni delivered the keynote at the May 5 symposium, Stanford’s first event uniting faculty and students with complementary backgrounds crucial for interdisciplinary research. This collaboration is intended to drive significant advancements in drug development, leveraging the combined efforts of industry, academia, and government to tackle challenges and seize opportunities.
Photos by Ramin Karchagani
Advancing Precision Health
Syliva Plevritis, professor of biomedical data science and of radiology, and chair of the Department of Biomedical Data Science (DBDS), noted the inaugural symposium complements the department’s mission of advancing precision health by applying computational methods, including AI and machine learning, to complex biomedical data sets.
“Where are we going?” asked Plevritis. “I believe that we’re in an intertwined revolution of AI and biotech. And this revolution that we’re experiencing together is manifesting itself as a knowledge revolution—a knowledge revolution on the molecular basis of health and disease. And we’re realizing that now by leveraging multimodal data.”
Plevritis said patients generate an abundance of biological information through their electronic health records, pathology, radiology, and genomics.
“At large academic medical centers like Stanford, we have bio specimen cores where we can generate research data on patients that are seen in our hospital and clinics,” she said. “And that integration of the research, deep molecular research data with the real-world data on the continuum of care and outcomes for patients is where I think the most exciting interface is for AI to really help us understand the biology of disease.”
Plevritis noted that the department is now 10 years old and growing, due in part by donors like the Warren Alpert Foundation, which hosted the symposium and gave the department a $5 million grant to fund 15 graduate and postdoctoral scholars in computational biology and AI. The foundation devoted to improving the health of the public has partnered with the department to train future scientists and leaders who can bridge two worlds: understanding both the biology of diseases and the AI methodology to study and potentially treat and cure them.
Research Spotlights: Quick Hits and Deep Dives
The symposium included a round of lightning talks by postdoctoral researchers from DBDS and other Stanford departments, as well as longer research talks by faculty from Stanford, UCLA and UCSF.
Edric Tam, the inaugural Warren Alpert Fellow in AI Computational Biology, gave a quick presentation on the statistical tradeoffs in deep generative models and synthetic data for biomedical applications. He highlighted two directions in his research. The first is when AI models try to learn from complex medical data, they always involve accuracy tradeoffs; he examines those tradeoffs and how to measure them. Tam then looks at what those tradeoffs mean in practice and whether we can trust the results.
In his research talk, Sohrob Shah, chief of computational oncology in the Department of Epidemiology and Biostatics at Memorial Sloan Kettering Cancer Center, highlighted the cancer center’s data science initiative, which has sequenced over 100,000 tumors and scanned 12 million pathology slides. He said a transformer model—a type of neural network that learns context and relationships using attention mechanisms—was fine-tuned to predict 124 cancer types. This is particularly important, Shah said, for the 5% of cancer patients whose tumors are undiagnosable, making it difficult for clinicians to treat.
“Scale is essential for granularity—and granularity is essential in diagnosis,” Shah said.
Olivier Gevaert, PhD, associate professor of medicine (computational medicine) at Stanford Medicine and of biomedical data science, runs a lab that focuses on multimodal data fusion, particularly in oncology and neuroscience, using various data types like images, text, and genomics data. Their goal is to combine digital pathology and genetic expression data to predict treatment responses in non-small cell lung cancer, which constitutes more than 80% of lung cancer cases and remains a leading cause of cancer-related mortality worldwide.
“We are very interested in mapping spatial omics to H&E images, because these are images that are routinely collected in clinic and they are the cornerstone of diagnosis in cancer patients. Fusion of different types of data likely will improve the performance,” Gevaert said. “And we have ongoing validation of these biomarkers in large, multimodal clinical trials of immunotherapy-treated patients in lung, breast and colorectal cancer.”
Industry Voices
A highlight of the day was an industry panel of biotech experts who spoke frankly about the transformative impacts of AI on various aspects of health care and drug development. For example, Tempus AI Inc. uses LLMs to interrogate electronic medical records (EMRs) for decision support and drug development. AstraZeneca focuses on oncology strategy and biometrics, leveraging AI to accelerate innovation.
The panel said big challenges still include AI’s ability to predict patient responses and the need for better data annotation. They highlighted the need for rigorous validation and the potential for AI to enhance, not replace, human expertise—with one panelist emphasizing the importance of critical thinking, focus and the ability to connect concepts.
Ezra Cohen, MD, however, took a contrarian view. The chief medical officer of oncology at Tempus AI Inc., believes AI will one day take the lead in medical care, and “the human is no longer in the loop.” He said the technology is nearly there and that AI has already proven to be more empathetic than human doctors and responds more accurately than most.
“AI follows guidelines with fidelity, so it’s not that big of a leap to have the AI write the progress notes and the management plan, and then go into the EMR and execute that plan,” Cohen said. “That last step, the world is not ready for. But I think it will happen.”
Alexander Morgan, a partner at Khosla Ventures and an alumnus of the PhD program in the Department of Biomedical Data Science, who moderated the industry panel, brought up that some biomedical data science trainees worry that AI will overtake their own talents and skills. He asked the panel of industry experts what they’re looking for in hiring.
Several panelists said they want recruits who can conduct rigorous validation of agentic systems and understand that AI is there to enhance, not replace, human expertise.
“Always be curious, always know what questions to ask,” said Judy Li, executive director of oncology biometrics at AstraZeneca. “The people who learn the most and grow the fastest are the people who ask the best questions.”
For Emily Fox, chief technical advisor at Insitro and professor of statistics and computer science at Stanford—it’s all about critical thinking and questioning AI results.
“For me, a focus is still on critical thinking skills,” Fox said. “You have to pick and pick and pick and be like, wait, that doesn’t make sense? You have to digest and analyze what these systems are producing and say, does that make sense biologically, does that make sense mathematically, statistically—and not just take the beautiful story.”
Student Voices
A closing poster session gave attendees a firsthand look at the work of trainees from Stanford, UCSF and the Gladstone Institutes are pursuing. Nineteen students—from biomedical data science to chemical engineering, genetics and cancer biology majors—presented poster abstracts on everything from characterizing pharmacogenetic variation among African ancestry participants in the NIH All of Us Research Program, to a Stanford pathology student examining whether mouse lymph node colonization models predict human tumor metastasis in melanoma.
Justin Adjasu, a DBDS Warren Alpert CBAI Scholar, presented: Deep Learning Analysis from Live-Cell Imagining of Cancer vs. TCR T Cell Interactions. The second-year master’s student showed how his research team is employing modern machine learning and AI tools to improve the analysis of how microorganisms behave together. They hope their approach will help fill the gaps in current methods and provide a clearer view of cell behavior.
The inaugural Computational Biology and AI Research Symposium made clear that the convergence of AI and biotechnology is no longer a distant promise—it is already reshaping how scientists understand disease, develop drugs, and care for patients. From foundation models that can diagnose rare cancers to multimodal data fusion that predicts treatment responses, the work on display reflected both the remarkable progress and the enormous challenges ahead. Participant feedback was excellent overall and the department looks forward to the next CBAI conference next spring.
Plevritis, William M. Hume Professor in the School of Medicine, is being honored for her pioneering work in computational cancer biology. The society has cited her algorithmic contributions as having defined how the field integrates multi-omics, imaging, and clinical data, revealing the biological foundations of the tumor microenvironment and its relationship to patient outcomes. The society also said her work as chair of Biomedical Informatics Graduate Program and as inaugural associate director for Cancer AI has shaped the science and institutional infrastructure of the field.
“A devoted mentor, she has trained over 60 graduate-level researchers, more than 20 of whom are now faculty at leading universities.”
The ISCB will honor the new fellows at the ISMB 2026 conference in Washington, D.C., this July.
In 2026, AI is no longer adjacent to healthcare—it is integral to it. AI is creating new opportunities to interpret complex biomedical data and generate insights relevant to research and patient care. But the human-machine relationship continues to evolve at breakneck speed—with an urgent need for guidance and support from all sectors. That includes research institutions, healthcare systems, industry partners, government agencies, and nonprofit organizations.
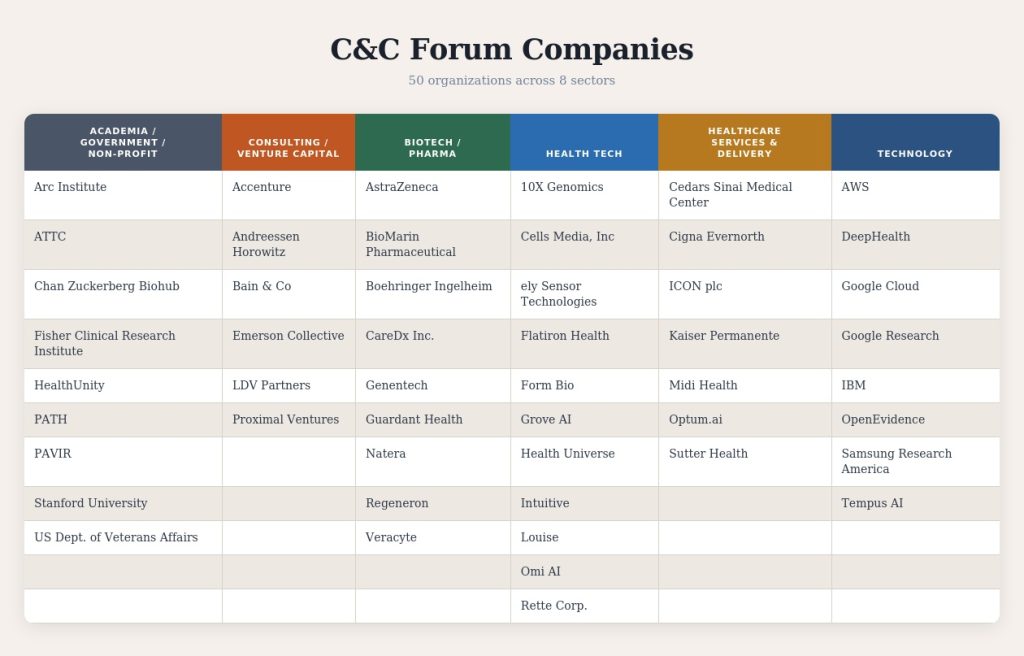
Stanford’s Department of Biomedical Science (DBDS) is at the center of this transformation. DBDS convened these many voices at its fourth annual Collaboration & Careers Forum (C&C) held January 27, 2026, at Stanford’s Arrillaga Alumni Center.
Enthusiasm showed up in the numbers: attendance rose more than 30 percent since last year, and company participation expanded from 40 to 52 organizations.
C&C attendees included senior leaders from global technology companies, venture capital firms, biotech and pharmaceutical companies, healthcare systems, government agencies and nonprofit research institutes. (Designed with the assistance of Claude (Anthropic)
“Multisector involvement will address the many needs of the AI-driven healthcare revolution that is already underway,” said Karen Matthys, MBA, Executive Director of DBDS Graduate Program & Strategic Initiatives and the driving force behind C&C. “These alliances – within Stanford, across Silicon Valley, and globally—don’t necessarily happen on their own but are essential for successful development and responsible implementation of AI-driven healthcare.”
Research brings methodological rigor, novel scientific insights, and long-term vision. Industry brings scale, infrastructure, pathways to real-world deployment, and resources for developing and scaling state-of-the-art technologies. Government agencies and nonprofit organizations are critical to provide not just funding support, but also a rich variety of data sources, important insights on AI guardrails, and valuable complementary perspectives. The result is a virtuous cycle: accelerating discovery while ensuring AI tools are validated, safe, and clinically meaningful.
Professor and Chair Dr. Sylvia Plevritis describes a nascent AI-assisted tumor board that employs a multimodal foundation model to guide precision cancer care.
Making Multimodal Data Actionable through AI
Multimodal data integration is increasingly central to precision health – and it is a main driver of DBDS research. Rich datasets that contain electronic health records (EHRs), genomic information, radiology and pathology images, and data from wearable devices are being combined with advanced AI models to reveal patterns unseeable by humans, traditional microscopes, and even the most state-of-the-art imaging platforms.
In her talk, Professor and Chair Sylvia Plevritis, PhD, highlighted challenges with complex, large datasets such as pathology data. To counter these issues, her lab is developing spatial biology tools and pathology foundation models to view and analyze pathology data as unique cellular ecosystems.
Linking diverse health data streams through multimodal foundation models has the potential to unearth deep biological insights that could refine diagnoses, predict disease trajectories, and tailor precision therapies based on a person’s unique characteristics and health history—across the continuum of care.
“EHRs are actually multimodal data timelines,” said Assistant Professor Jason Fries, PhD. In his research talk, Fries described the challenge of constructing reliable longitudinal patient trajectories from noisy, biased, and incomplete clinical data – and the critical need for improved feedback loops to support effective human-AI teaming.
Data collected from wearables like smart watches and Oura rings can provide a wealth of information about women’s health. Professor Barbara Engelhardt, PhD, presented her current study, “Study on Typically Ignored Groups of Menstruating Adults (STIGMA),” which has collected wearable data for a year on 304 menstruating people and is developing AI tools to analyze the complex data.
Across C&C talks and discussion, one key truth came to light: the urgent need for robust frameworks to evaluate AI systems. While advances in model accuracy have been impressive, faculty continually emphasized that accuracy alone is insufficient.
How should AI safety be measured? How can performance be monitored over time in dynamic clinical environments? What early warning indicators might serve as a “canary in the coal mine” if an AI model begins to drift or fail?
Jonathan Chen and his lab are asking critical questions like: Can an AI model function safely and effectively within real clinical workflows?
Developing systematic approaches to assess reliability, robustness, and long-term effectiveness remains one of the field’s most pressing challenges, as noted by Associate Professor Jonathan Chen, PhD, in his talk.
Echoing this notion, during the event’s afternoon industry panel, speakers stressed that successful implementation of AI in healthcare depends not only on sophisticated models, but on trust, transparency, and ongoing verification. The panel, “Humans, Agents, & Infrastructure: The Future of AI in Health,” was co-moderated by DBDS students Susie Avagyan (PhD candidate) and Shlok Natarajan (MS candidate). Panelists included Dr. Amar Das, Vice President, Real World Evidence, Guardant Health; Dr. Jing Huang, Chief Data & AI Officer, CareDx, Inc.; and Devi Ramanan, Managing Director, Accenture.
Acknowledging real-world challenges of achieving precision health, Assistant Professor Alex Ioannidis, PhD, spoke about how broad labels like “Hispanic” or “Mexican” do not accurately reflect people’s genetic backgrounds, which can muddy research results. Most people in Mexico have mixed ancestry from Indigenous American populations, Europeans, and Africans (to a smaller extent) —which is reflected by small but meaningful differences in their DNA.
His research illuminates how important it is for DNA information to be extremely precise—toward understanding how different ancestries affect both health and response to medications. In a similar vein, Professor Teri Klein, PhD, spoke about her work to personalize treatment with medications, recognizing that people’s responses to drugs are different based on genetics. Klein’s research has helped create tools that guide doctors on the safest and most effective medication doses. Some of these guidelines are now recognized by the U.S. Food and Drug Administration and included on official drug labels.
Connecting People and Ideas
As a two-way exchange of information, energy, and new directions, the C&C enables DBDS faculty and trainees to explore various ways to work together. Each participating organization hosts a dedicated table, creating space for discussions about research partnerships, data sharing, advising relationships, and long-term collaborations.
As with previous C&C events, DBDS trainees were active participants to prepare for their role as future leaders. Graduate students and postdocs moderated sessions, engaged directly with external partners, and showcased the depth of talent emerging from DBDS training programs.
For trainees navigating a rapidly evolving AI landscape, the C&C offered a chance to watch how AI is being deployed in biomedicine today and where it is headed next.
As attendance and engagement continue to grow, the C&C has become more than an annual event. It reflects DBDS’s expanding role within the tech- and health-heavy Bay Area region and the broader biomedical AI community both nationally and internationally. Convening diverse stakeholders around shared research challenges shows how DBDS’ persistent leadership will facilitate making AI research and deployment both scientifically rigorous and clinically meaningful.
For more information about C&C, including videos and photos from the event, visit the DBDS 2026 C&C website.