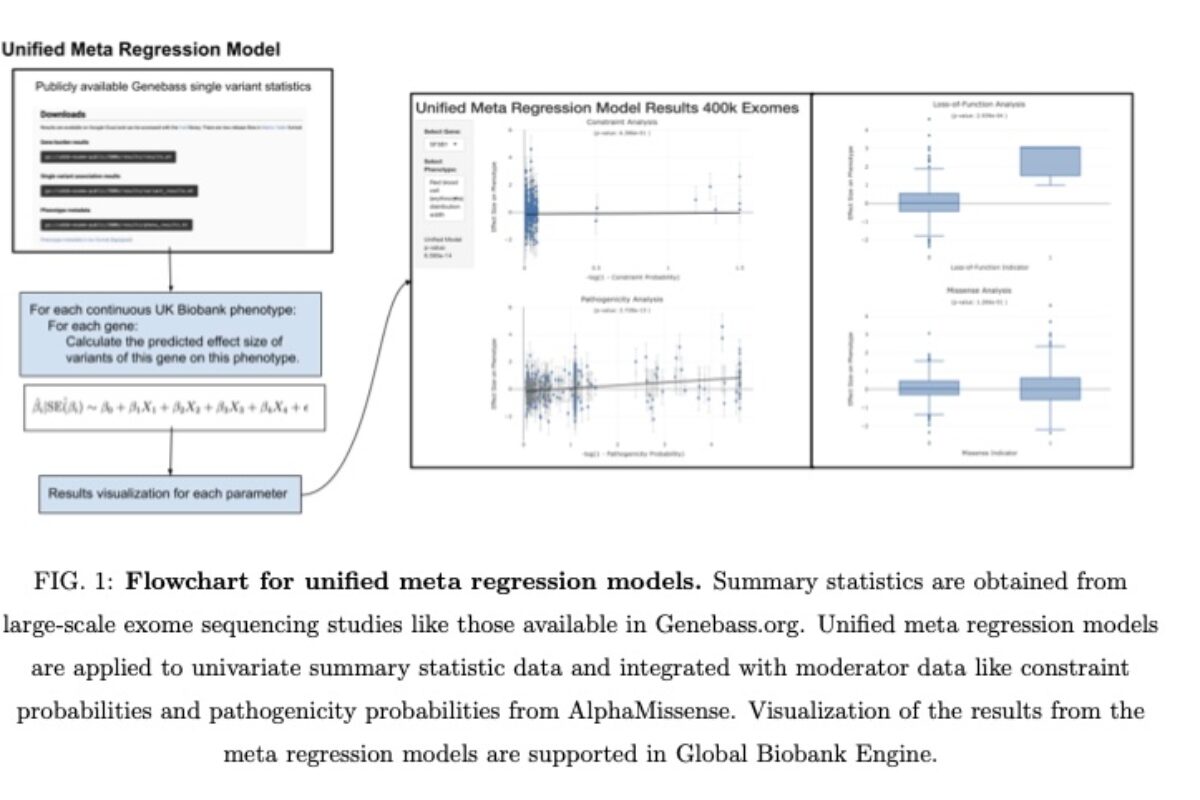
Rare variant association studies (RVAS) of complex traits have emerged as a powerful approach to advance drug discovery and diagnostics.
Read more here: https://www.biorxiv.org/content/10.1101/2025.01.23.634522v1
Rare variant association studies (RVAS) of complex traits have emerged as a powerful approach to advance drug discovery and diagnostics.
Read more here: https://www.biorxiv.org/content/10.1101/2025.01.23.634522v1

“Medicine is in the dawn of a fundamental shift from using artificial intelligence (AI) as tools to deploying AI as agents. When used as a tool, AI is passive and reactive. Even powerful medical AI foundation models today remain tools that depend on human users to provide input and context, interpret their output, and take follow-up steps. To fully unlock AI’s potential in medicine, clinicians need to make the key conceptual shift from using AI as sophisticated calculators to embracing AI as health-care teammates.”
Read more here: https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(25)00202-8/abstract

DBDS Student Talks (BIOMEDIN 201) Winter Quarter 2024-25 in Alway M114/ZOOM. Lunch will be provided for the talks.
Upcoming 2/24 Monday Talk Speakers:
| DATE | 12:15-12:45 | 12:45-1:15 |
| 2/24/2025 | Bedi, Suhana | Hassan, Sohaib |
Attendance will be taken weekly on a Google sheet. Please sign your name on the sheet when you attend the Monday Talks via Zoom or in-person.
–Hybrid model, with both in-person and remote attendance via Zoom.
–If you require any special accommodations, please reach out to DBDS Student Services in advance before the date of your talk.
Zoom Link: https://stanford.zoom.us/j/97615772092?pwd=J5LziKarW0NjoJ7QKHFKZMOX8lh2gS.1&from=addon
Password: 562775

“This is a new era for us,” Chair of the Department of Biomedical Data Science Sylvia Plevritis began, opening the third annual Collaboration & Careers Forum on January 16, 2025. The event, which connected leaders from various sectors with DBDS faculty, students, and researchers to discuss technological advancements in precision healthcare, focused heavily on new developments in AI. It explored how those advancements could be applied to improve efficiencies and health outcomes.
Read the story here: https://dbds.stanford.edu/collaboration-and-careers-2025-overview/https://dbds.stanford.edu/collaboration-and-careers-2025-overview/